phage genome assembly and annotation tool
Easy, Fast & Reliable
Phage Genome Assembly, Annotation & Genomic Comparison Software
Automated
Reliable
No Coding
Use for free


Features Scientists Rely On


We Are Serious About Our Science
Our academic expertise in phage bioinformatics has focused on phage-bacterium coevolution, pioneering new annotation methods, and investigating the uncharted territories of phage genomics.
IT infrastructure
Our state-of-the art pipelines are powered by Finnish hardware, delivering advanced phage genomics in minutes, not days!
development
We are committed to continuous learning, and are constantly developing our pipelines and algorithms to increase the accuracy of our tools.
From Raw Data to Ready-to-Use Results
Phagenomics Core accelerates your phage genome assembly, annotation, and comparison.
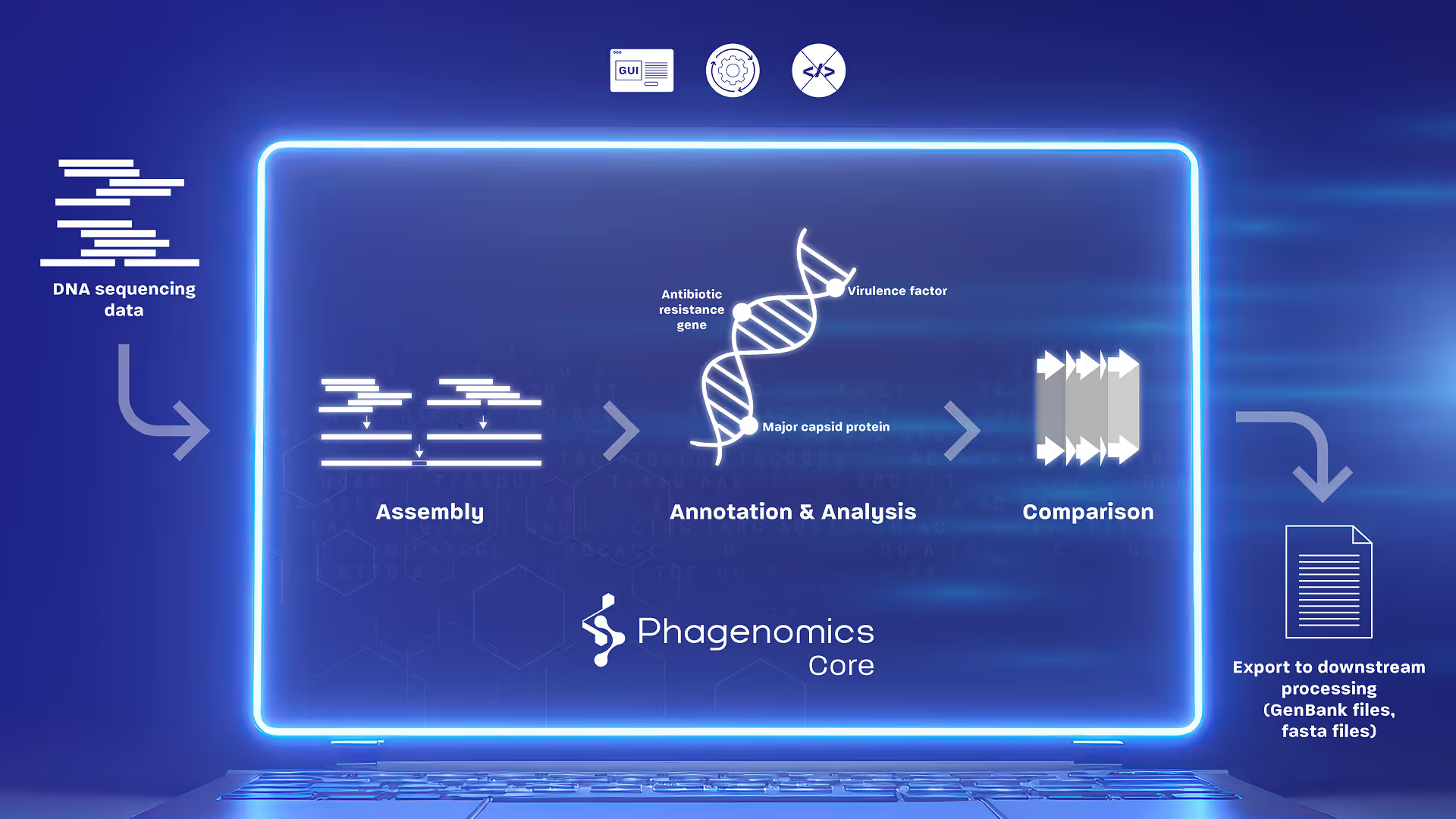
Phagenomics Core Combines All the Essential Tools
We utilize third-party and proprietary tools to deliver optimized results.
Assembly tools in Core
Pilon - Polishing genome with raw reads
Bowtie - Read mapping to contigs
SPAdes - Genome assembly
SAMtools - SAM file conversion, sorting and indexing
BlobTools - Characterisation of assembled contigs
Trimmomatic - Read trimming, pairing and quality control
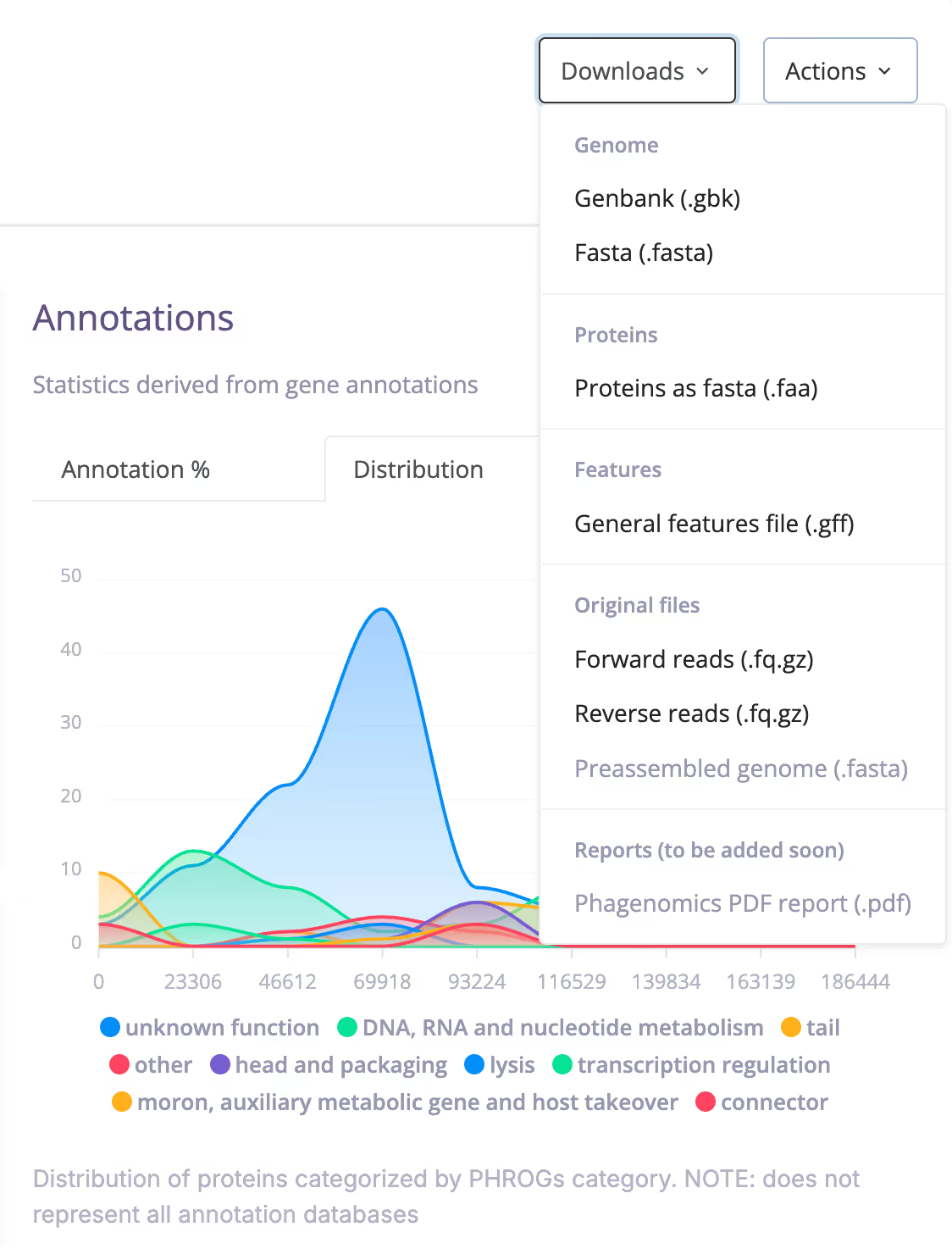
Annotation & Analysis tools in Core
Pyani - Generate heatmaps based on genome alignments
CheckV - Measuring phage genome completeness
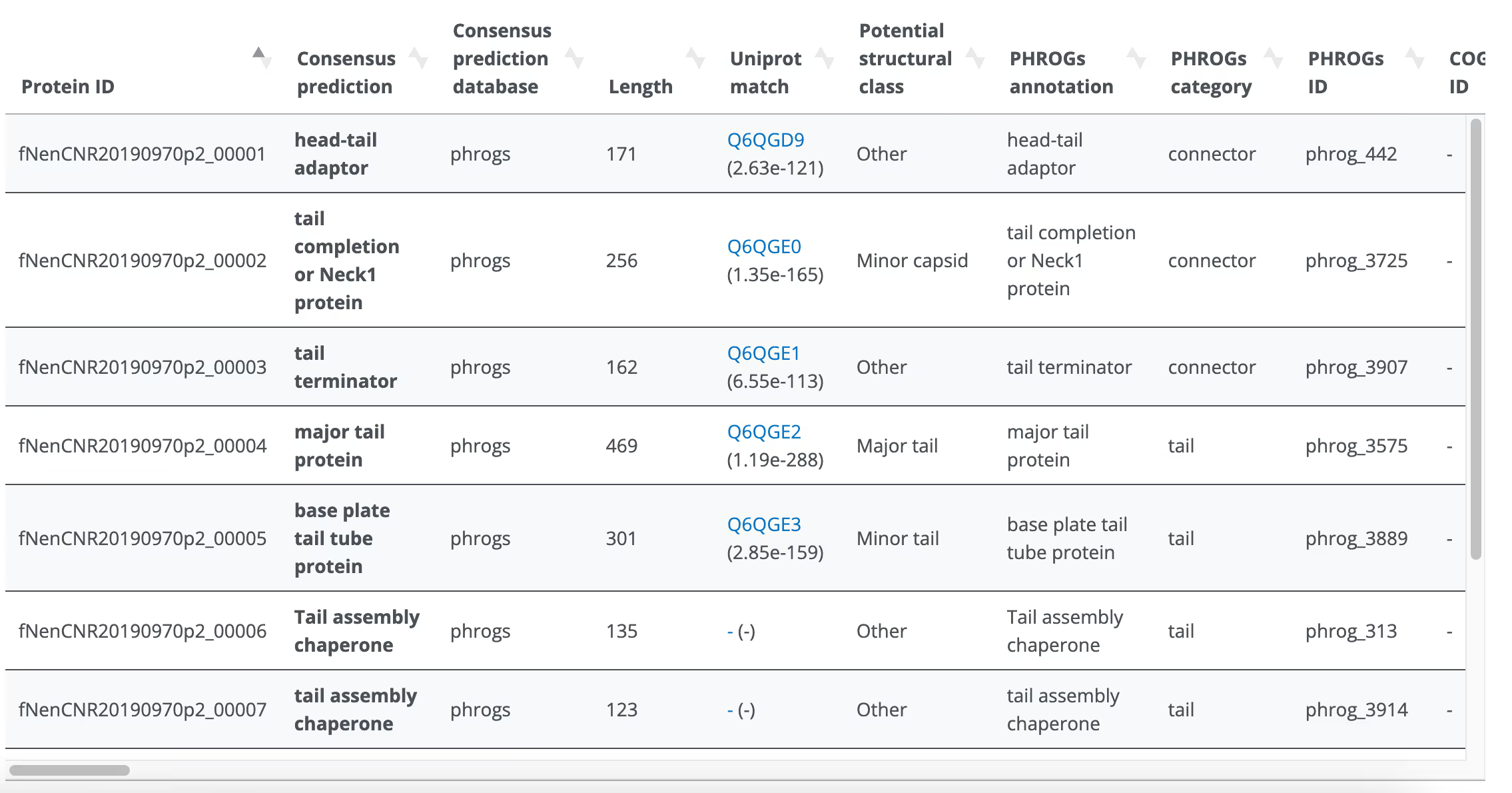
PhANNs - Structural protein prediction
Prodigal - ORF prediction
Prokka - Initial annotation
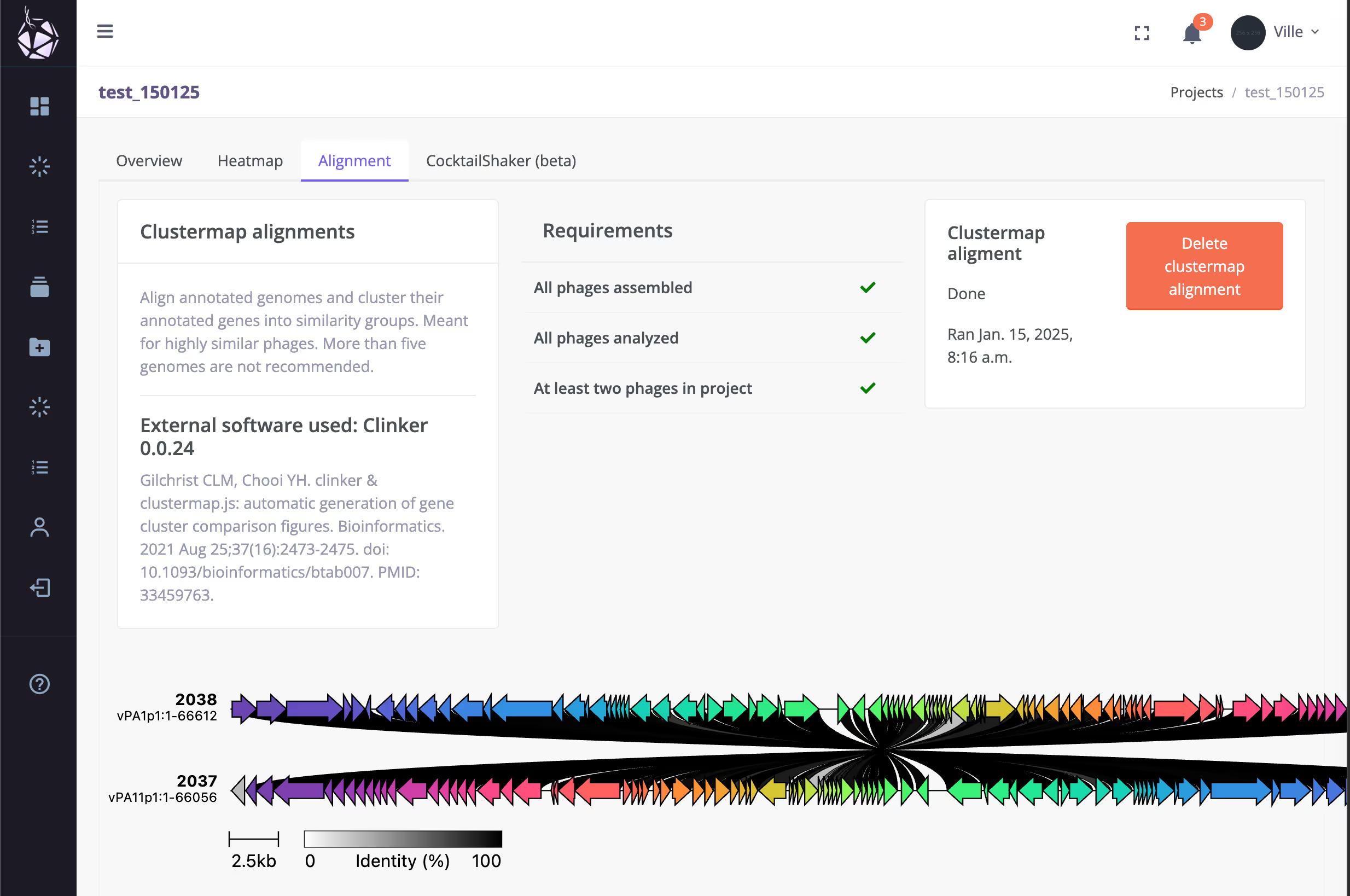
Clinker - Align and visualise whole genomes
Bacphlip - Phage lifestyle prediction
PhageTerm - Predicting phage genome type and genome ends
AMRFinderPlus - Antibiotic resistance gene prediction
VirulenceFinder - Finding virulence genes
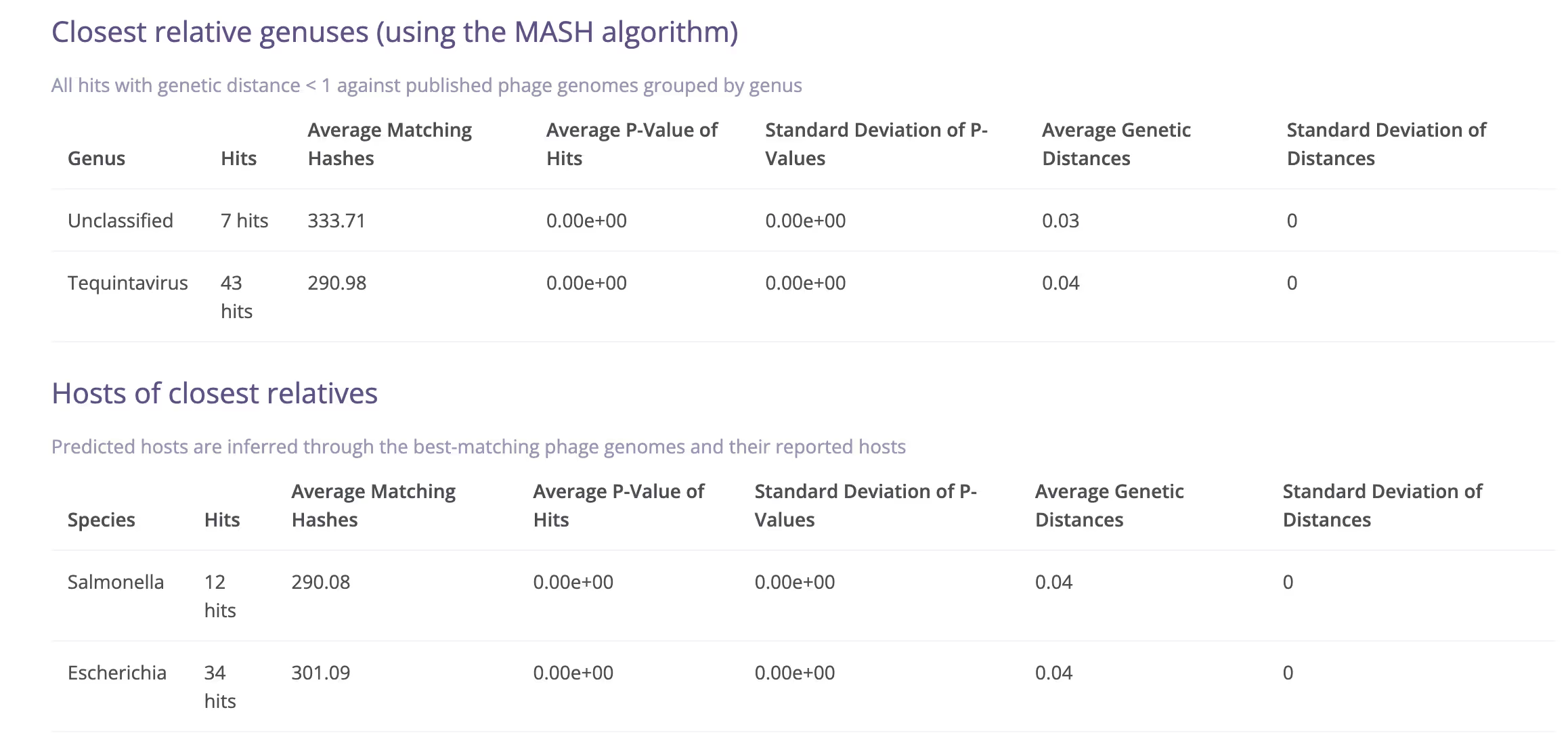
The Millardlab database - Matching your phage sequences

What Scientists Say
Our roots

Phagenomics Pricing
Flexible plans for people pushing the boundaries of genomic science.

Fast results
Phagenomics is developed by PrecisionPhage, a company dedicated to furthering groundbreaking research and innovative solutions that drive success in biotechnology. Our commitment to excellence has established us as a trusted partner in the industry.
Average time to assemble
Average time to annotate
FAQs
If your question is not listed below, don’t hesitate to contact us!
It is true that genome assembly and analysis is generally composed of automated steps (e.g. assembling a genome) and lots of manual annotation and manual quality control. We used to do everything by hand too, but quickly noticed the redundancy in our actions – and it is from this lack of automation that Phagenomics was born.In automating the assembly and annotation processes, we aim to capture the manual actions of a researcher on an algorithmic level while drastically cutting the time used for phage analysis. This approach also enables a standardised and reproducible set of methods for phage genomics – something we often see missing in published phage genomes. For example, one crucial step in assembly is to fish out the main phage contig from all the contigs output by an assembler. A researcher would manually do this by mapping raw reads back onto the contigs and observe the read coverage distribution as well as contig sizes and GC%. Finally, they would choose the high coverage contig that represents the desired amplified phage genome (and if several contigs were of high coverage, they would likely abandon the problematic sequencing project and resequence the phage). With Phagenomics we mimic this behaviour, automatically picking the high coverage phage contig from the sea of mostly bacterial contigs (we do give you the option of manually choosing another contig too). In case of multiple competing contigs or the apparent lack of viral sequences, we warn the user not to continue further. We use similar steps in other parts of the pipeline too, for example in phage genome end detection (see separate FAQ for that).However, if you are hesitant with a fully automated approach, you can always use Phagenomics as a starting point and do any manual adjustments later using the provided fasta, genbank (.gbk) and general features file (.gff) of an analysed phage.
This is a tricky one. Phage genomes vary in their genome types and replication strategies and we've spent a long time optimising our system to accommodate common genomes as well as edge cases. We begin by finding common assembly artefacts in the initial assembled genome. After fixing these via proprietary algorithms, we examine the genome using PhageTerm to determine replication types. We also look for common signature genes from which genomes are conventionally set to start from. Concatenating these data, we then use custom algorithms to flip the genome from an optimal position. In case of terminally redundant genomes, redundant ends are placed at the 5' and 3' ends of the genome. In case of cohesive ends, we flip the genome to start from the cohesive end (in case of 5' end) or end with the cohesive end (in case of 3' end). We do not use PhageTerm outputs as is, as they have been found to produce artefacts in our results. Note that for full certainty of physical genome ends, experimental verification using for example Sanger sequencing is the gold standard. You can always assemble your genome using Phagenomics and then design primers for genome end verification. After experimental verification, you could rearrange the genome using your software of choice and then upload your modified genome as a "preassembled" genome for analysis in Phagenomics (we do not reorient preassembled genomes). Or you can outsource all this to us: please contact us at customerservice@precisionphage.com.
Phagenomics was created by academics and we fully appreciate the need to understand the methods used for assembling and analysing your phage genome. Our tools work through a pipeline of interlinked scripts. These scripts are either our own proprietary code or "wrappers" for 3rd party open source programs (used within the bounds of their respective licenses). When you finish a phage analysis, a list of citations against these 3rd party programs is generated. This list contains version numbers, citation information and explanation of how the tool was used in the analysis of your phage, so you will have understanding of what was actually done. As a commercial actor, we cannot disclose the details of our own proprietary algorithms, but still aim to provide you with an overview of what was done. We are currently implementing details of the procedure in the Materials and methods section of a finished analysis, but please contact us at customerservice@precisionphage.com.
All payments made through Phagenomics are processed using Stripe, a leading global payment processor trusted by millions of businesses worldwide. Stripe is fully PCI compliant and uses advanced encryption methods to ensure your financial data is always securely transmitted and stored. Your credit card information is never stored on our servers and is completely handled by Stripe.
Unfortunately, we currently only support assembly from Illumina paired end raw reads. However, you can always upload and analyse a preassembled genome that was based on NanoPore or Pacbio sequencing.
The "Materials and methods" tab on an analysed phage's page lists all third party tools used in the analysis (and assembly if applicable) and their version and citation information. We also report the version of Phagenomics used in the analysis. We encourage researchers to cite Phagenomics and 3rd party tools, when they are used in your study. For Phagenomics, cite "Phagenomics phage genome analysis portal" and add date of use.
For raw reads, we currently only accept Illumina paired end reads in FastQ format. The files may be gzipped or not. Alternatively, you may upload a preassembled genome (.fasta file) that was made using any technology.
We can certainly accommodate batch processing of multiple genomes using our batch processing system. However, this system is not yet available on the web service. Please contact us at customerservice@precisionphage.com for a custom solution.
Yes, you will retain ownership of your data after upload. However, do note that we may use your phage data for training AI models in the future. Please review our policies for further information.
Your data is securely stored using DigitalOcean's Object Storage, which is designed for durability and security. Importantly, all files stored in our system are set to 'Private' by default. This means files are not publicly accessible and can only be accessed through secure authentication protocols established by our system. Please remember that while we implement comprehensive security measures, no method of transmission or storage is 100% secure. We highly encourage users to employ security best practices on their end too, such as using strong and unique passwords.
Usually between 8 to 15 minutes, depending on the size and complexity of the genome.
Unless you cancel your subscription before the trial ends, the subscription will automatically renew. If you cancel in the middle of your trial, you will still have access to PRO features until the end of the trial.
Phagenomics is the only comprehensive and user-friendly phage genome assembly, analysis and comparison web portal. While many of the tools we use in our pipelines are open source and can be used by experts with command line and bioinformatics know-how, we bundle them all into a single streamlined package. On top of this we've developed our own tools and quality control checkpoints, which result in higher quality genomic assemblies and annotations.We are currently benchmarking the system against published genomes, stay tuned.
In Phagenomics, "analysis" is what follows "assembly". The analysis pipeline is a complicated set of rules that scrutinise the assembled genome to extract as much data out of it as possible. We begin by polishing your phage genome with raw reads (if available) to ensure no sequencing errors remain. We then determine physical genome ends (see separate FAQ on this). After obtaining the optimal continuous contig, we predict ORFs using Prodigal and employ several HMM profiles and to annotate each ORFs using custom scripts. We also use AI-based 3rd party software to predict structural protein coding genes. All annotations are combined into one master annotation that works through hierarchical concatenation of several annotation inputs. In addition to gene predictions, we run multiple proprietary and 3rd party tools to characterise the genome in terms of completeness, virulence, antibiotic resistance and relatedness to existing phage genomes.
Accelerate Your
Bioinformatics Today